
Cystic fibrosis (CF) is the most common fatal autosomal recessive disease among the Caucasian population. An individual must inherit a defective copy of the CF gene (one from each parent) to have CF. Cystic fibrosis is usually diagnosed in infancy or early childhood, but patients may be diagnosed later in life. For individuals diagnosed later in life, respiratory symptoms are frequently the major manifestation of the disease.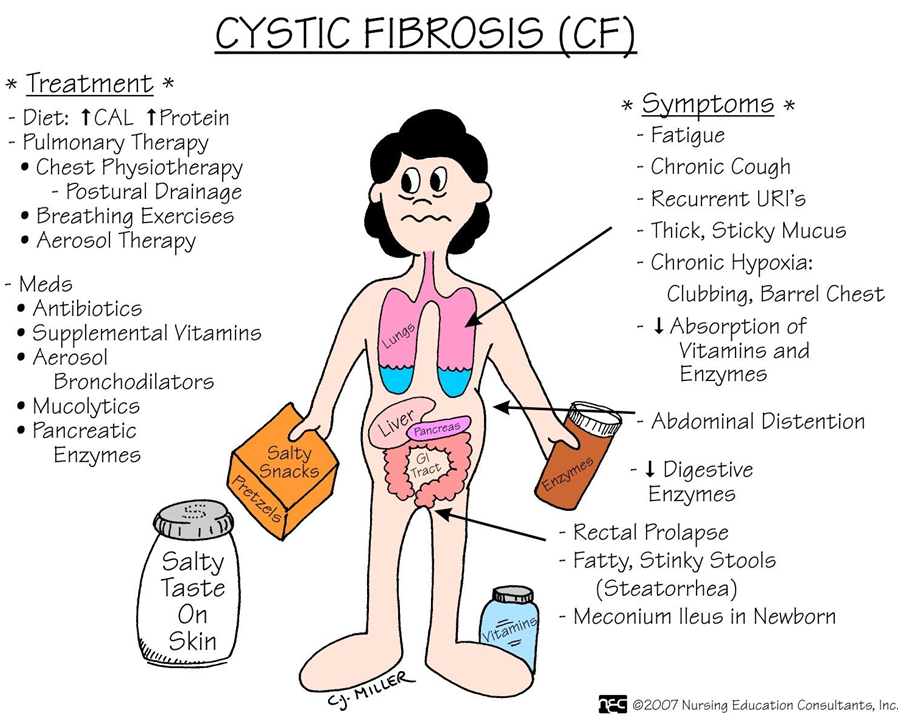
Pathophysiology
This disease is caused by mutations in the CF transmembrane conductance regulator protein, which is a chloride channel found in all exocrine tissues. Chloride transport problems lead to thick, viscous secretions in the lungs, pancreas, liver, intestine, and reproductive tract as well as increased salt content in sweat gland secretions. In 1989, major breakthroughs were made in this disease with the identification of the CF gene. The ability to detect the common mutations of this gene allows for routine screening for this disease as well as the detection of carriers. Genetic counseling is an important part of health care for couples at risk.
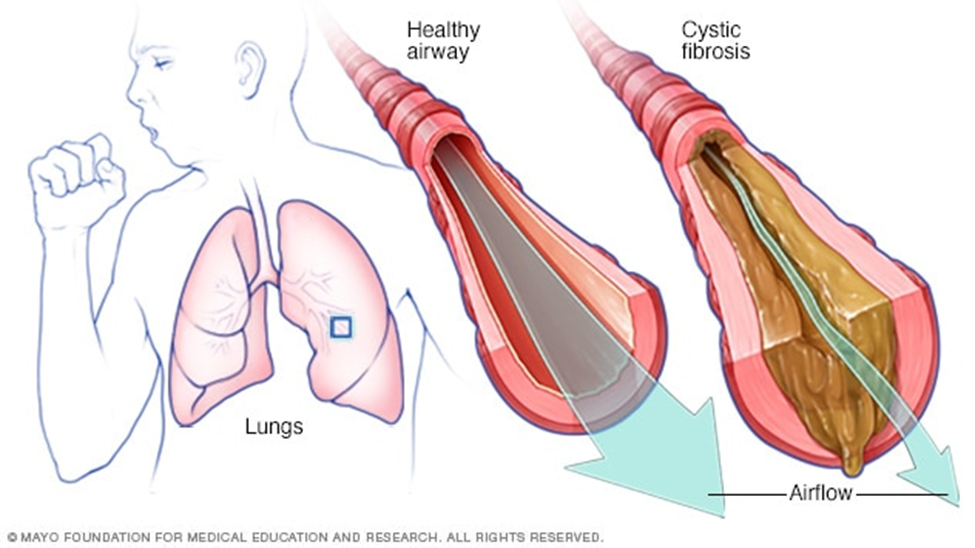
Airflow obstruction is a key feature in the presentation of CF. This obstruction is due to bronchial plugging by purulent secretions, bronchial wall thickening due to inflammation, and, over time, airway destruction. These chronic retained secretions in the airways set up an excellent reservoir for continued bronchial infections.
Clinical Manifestations
The pulmonary manifestations of this disease include;
- a productive cough, wheezing, hyperinflation of the lung fields on chest x-ray, and pulmonary function test results consistent with obstructive airways disease.
- Colonization of the airways with pathogenic bacteria usually occurs early in life.
- Staphylococcus aureus and Haemophilus influenzae are common organisms during early childhood. As the disease progresses, Pseudomonas aeruginosa is ultimately isolated from the sputum of most patients.
- Upper respiratory manifestations of the disease include sinusitis and nasal polyps.
Nonpulmonary clinical manifestations include;
- gastrointestinal problems (eg, pancreatic insufficiency, recurrent abdominal pain, biliary cirrhosis, vitamin deficiencies, recurrent pancreatitis, weight loss),
- genitourinary problems (male and female infertility), and
- Clubbing of the extremities.
Assessment and Diagnostic Findings
Most of the time, the diagnosis of CF is made based on an elevated result of a sweat chloride concentration test, along with clinical signs and symptoms consistent with the disease. Repeated sweat chloride values of greater than 60 mEq/L distinguish most individuals with CF from those with other obstructive diseases. A molecular diagnosis may also be used in evaluating common genetic mutations of the CF gene.
Medical Management
Pulmonary problems remain the leading cause of morbidity and mortality in CF. Because chronic bacterial infection of the airways occurs in individuals with CF, control of infections is key in the treatment.
- Antibiotic medications are routinely prescribed for acute pulmonary exacerbations of the disease. Depending upon the severity of the exacerbation, aerosolized, oral, or intravenous antibiotic therapy may be used. Antibiotic agents are selected based upon the results of a sputum culture and sensitivity. Patients with CF have problems with bacteria that are resistant to multiple drugs and require multiple courses of antibiotic agents over long periods of time.
- Bronchodilators are frequently administered to decrease airway obstruction. Differing pulmonary techniques are used to enhance secretion clearance. Examples include manual postural drainage and chest physical therapy, high-frequency chest wall oscillation, and other devices that assist in airway clearance (PEP masks [masks that generate positive expiratory pressure], “flutter devices” [devices that provide an oscillatory expiratory pressure pattern with positive expiratory pressure and assist with expectoration of secretions]).
- Inhaled mucolytic agents such as dornase alfa (Pulmozyme) or N-acetylcysteine (Mucomyst) may also be used. These agents help to decrease the viscosity of the sputum and promote expectoration of secretions.
- Anti-inflammatory: To decrease the inflammation and ongoing destruction of the airways, anti-inflammatory agents may also be used. These may include inhaled corticosteroids or systemic therapy. Other anti-inflammatory medications have also been studied in CF. Ibuprofen was studied in children with CF and some benefit was demonstrated, but there is little information on its use in young or older adults with CF.
- Supplemental oxygen is used to treat the progressive hypoxemia that occurs with CF. It helps to correct the hypoxemia and may minimize the complications seen with chronic hypoxemia (pulmonary hypertension).
- Lung transplantation is an option for a small, select population of CF patients. A double lung transplant technique is used due to the chronically infected state of the lungs seen in end-stage CF. Because there is a long waiting list for lung transplant recipients, many patients die while awaiting a transplant.
- Gene therapy is a promising approach to management, with many clinical trials underway. It is hoped that various methods of administering gene therapy will carry healthy genes to the damaged cells and correct defective CF cells. Efforts are underway to develop innovative methods of delivering therapy to the CF cells of the airways.
Nursing Diagnosis
Based on the assessment data, the major nursing diagnoses are:
- Ineffective airway clearance related to thick, tenacious mucus production.
- Ineffective breathing pattern related to tracheobronchial obstruction.
- Risk for infection related to bacterial growth medium provided by pulmonary mucus and impaired body defenses.
- Imbalanced nutrition: less than body requirements related to impaired absorption of nutrients.
- Anxiety related to hospitalization.
- Compromised family coping related to child’s chronic illness and its demands on caregivers.
- Deficient knowledge of the caregiver related to illness, treatment, and home care.
Nursing Management
Nursing care of the adult with CF includes assisting the patient to manage pulmonary symptoms and to prevent complications of CF.
- Specific nursing measures include strategies that promote removal of pulmonary secretions; chest physiotherapy, including postural drainage, chest percussion, and vibration, and breathing exercises are implemented and are taught to the patient and to the family when the patient is very young.
- The patient is taught the early signs and symptoms of respiratory infection and disease progression that indicate the need to notify the primary health care provider.
- The nurse emphasizes the importance of an adequate fluid and dietary intake to promote removal of secretions and to ensure an adequate nutritional status. Because CF is a life-long disorder, patients often have learned to modify their daily activities to accommodate their symptoms and treatment modalities.
- Although gene therapy and double lung transplantation are promising therapies for CF, they are limited in availability and largely experimental. As a result, the life expectancy of adults with CF is shortened.
- For the patient whose disease is progressing and who is developing increasing hypoxemia, preferences for end-of-life care should be discussed, documented, and honored.
- Patients and family members need support as they face a shortened life span and an uncertain future.